paired end sequencing r1 and r2
Paired-End Reads There are two FastQ files generated in an Illumina paired-end reads sequencing run. R1139283692 R2137472495 This usually does not happen for PE sequencing which makes me a bit nervous to proceed with analysis.
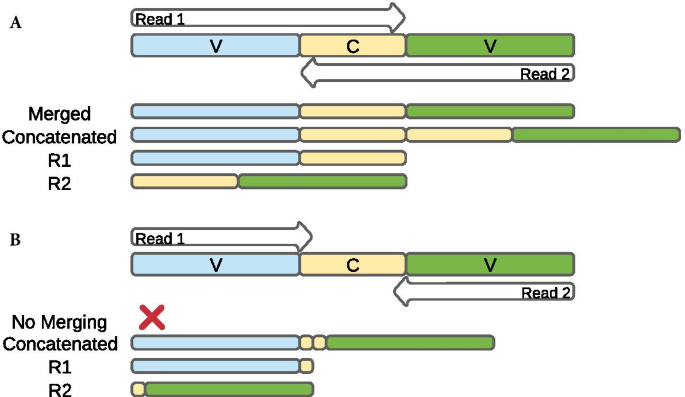
Concatenation Of Paired End Reads Improves Taxonomic Classification Of Amplicons For Profiling Microbial Communities Bmc Bioinformatics Full Text
For each dataset we used the miRNA module using paired-end mode and single end mode for the R1 and R2 reads.

. If the sequencing sample is the same as the original copy the read R1 should be mappable in forward direction in. The map is 80 but the R1 R2 counts are. For paired-end alignment aligners want the R1 and R2 fastq files to be in the same name order and be the same length.
We present the PEAR. The direction and positional order of the paired-end reads R1R2. For the paired-end mode we initially joined reads using two.
The files have this naming convention. The steps below can be used if. The reverse reads are sequenced in a reversed manner but the content of the reverse read is.
Therefore a robust tool is needed to merge paired-end reads that exhibit varying overlap lengths because of varying target fragment lengths. Merging paired end reads R1 and R2 files 10-09-2013 0730 AM. Analysis modes of NGmerge.
Normally R1 and R2 are not reverse complements of each other. So while R1 starts with your 5 adapter for R2 anything is possible depending on the length of the fragment of interest and the read length. To anyone who may have dealt with Illumina MiSeq paired end reads.
Normally R1 and R2 are not reverse complements of each other. First sequencing can either be single-end where each sample has only sequence data or paired-end where each sample has two sequence data R1 and R2. The reverse reads are sequenced in a reversed manner but the content of the reverse read is.
For each cluster that passes filter a single sequence is written to the corresponding samples R1 FASTQ file and for a paired-end run a single sequence is also. What are the best programsscripts to. You could see the reverse.
Adapter trimming can remove FASTQ sequences if the. The diagrams show the paired-end reads R1 R2 derived from sequencing DNA fragments white boxes with sequencing adapters gray boxes.

Casava Files Of Paired End Reads General Discussion Qiime 2 Forum

Deal With Single End And Paired End Data Bioinformatics
Eager Nf Core

Epitranscriptomic Mapping Of Rna Modifications At Single Nucleotide Resolution Using Rhodamine Sequencing Rho Seq Star Protocols

How I Will Import Combined R1 Fastq And R2 Fastq Files Into Qiime2 User Support Qiime 2 Forum

Complementary Fastq File Compare With Original Rna Issue 1068 Alexdobin Star Github

Flow Diagram Outlining The Sequence Data Analysis Process Download Scientific Diagram

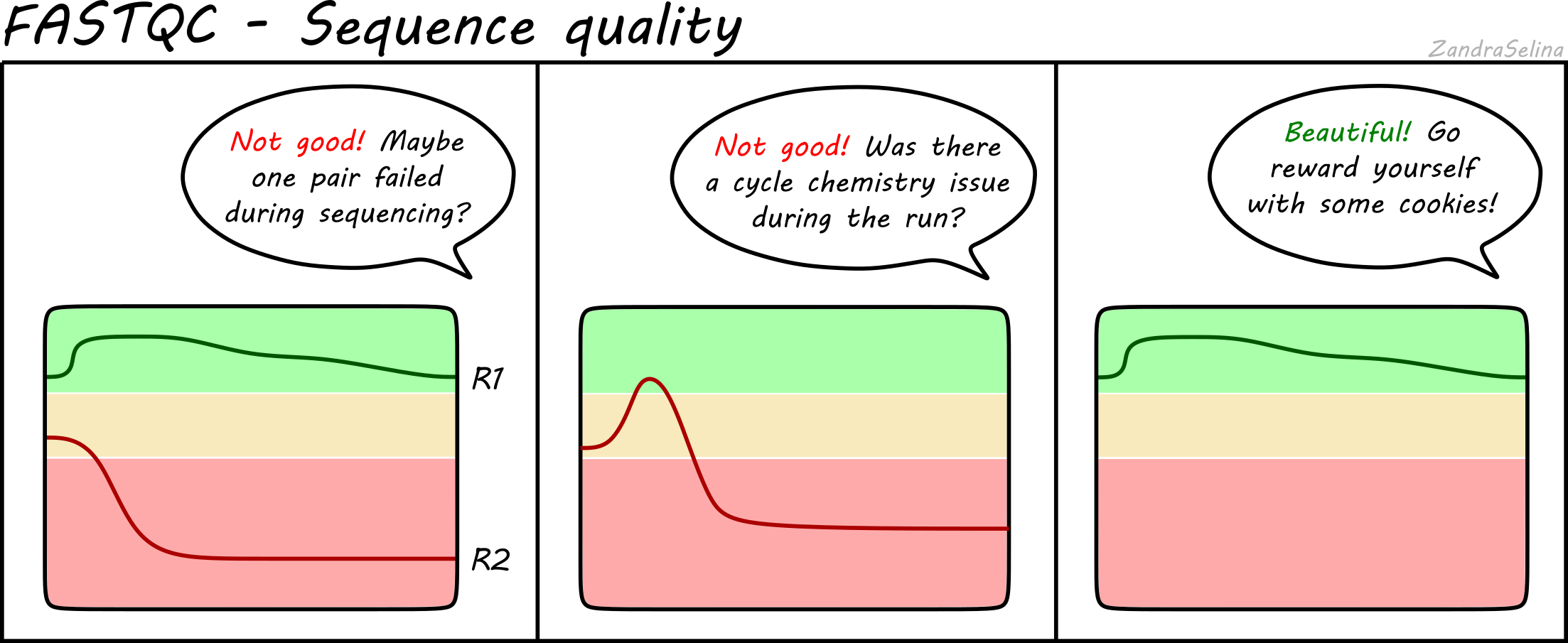
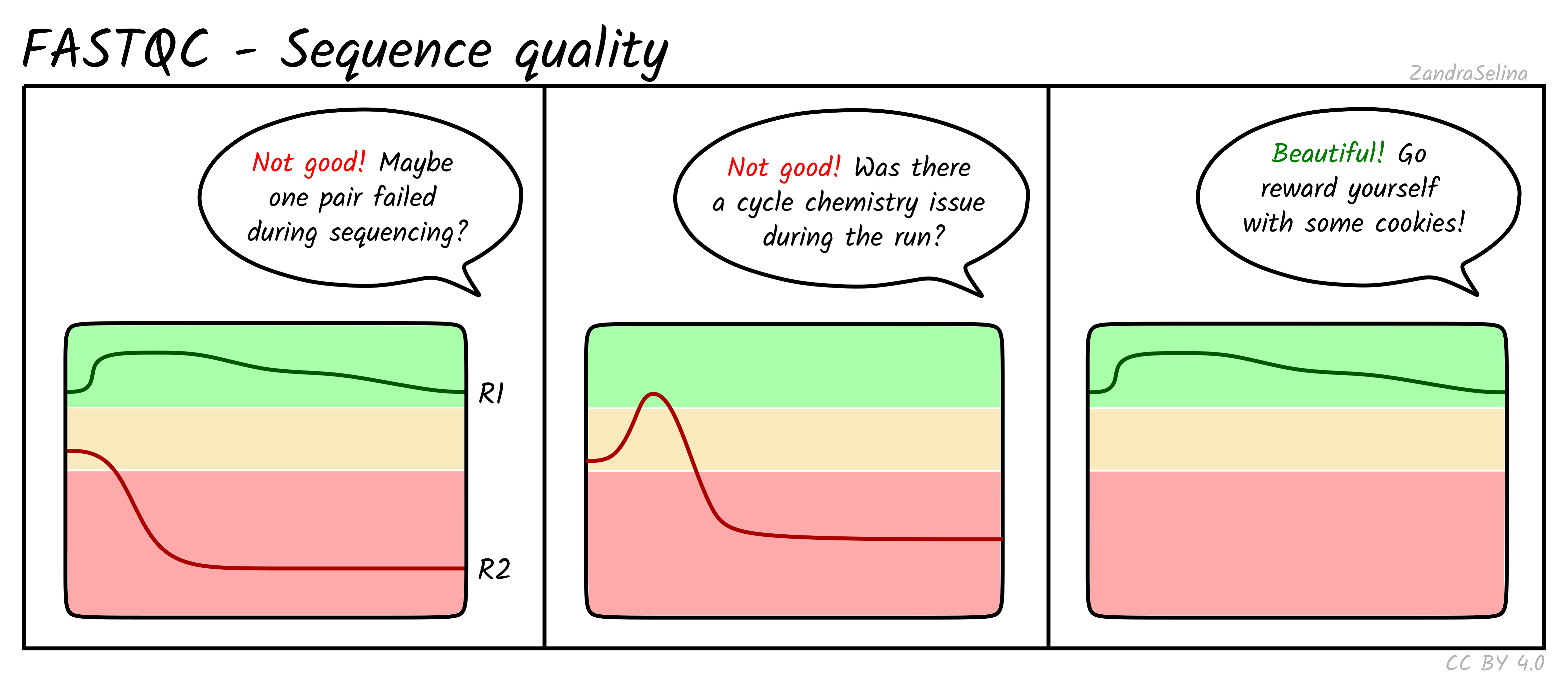
Long Fragments Achieve Lower Base Quality In Illumina Paired End Sequencing Abstract Europe Pmc

Multiplexed Primer Extension Sequencing Enables High Precision Detection Of Rare Splice Isoforms Biorxiv
Eager Nf Core
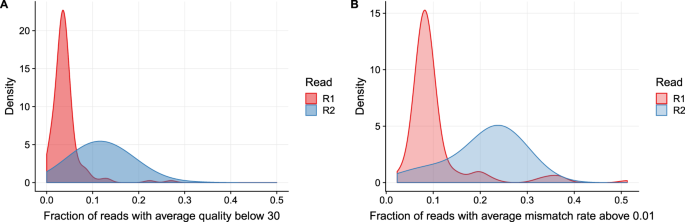
Long Fragments Achieve Lower Base Quality In Illumina Paired End Sequencing Scientific Reports
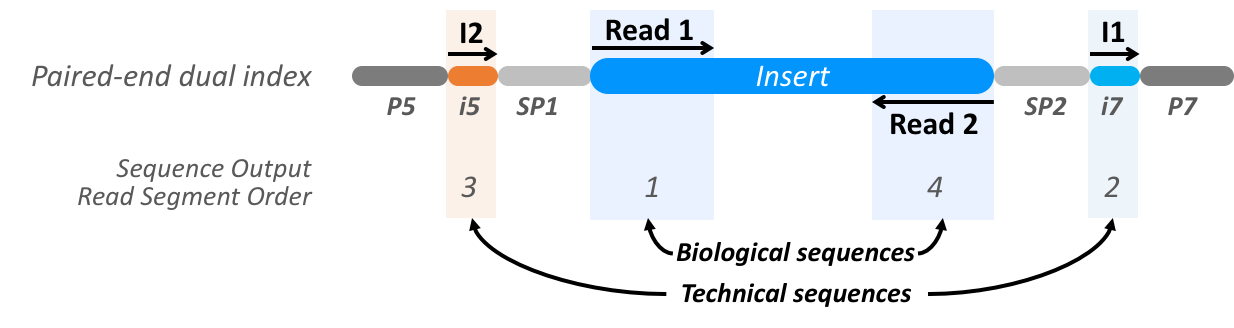
Read Segment Transformation In Pheniqs

Many 35nt Reads With N Bases In R1 Paired End When Sequencing With Illumina Nextseq
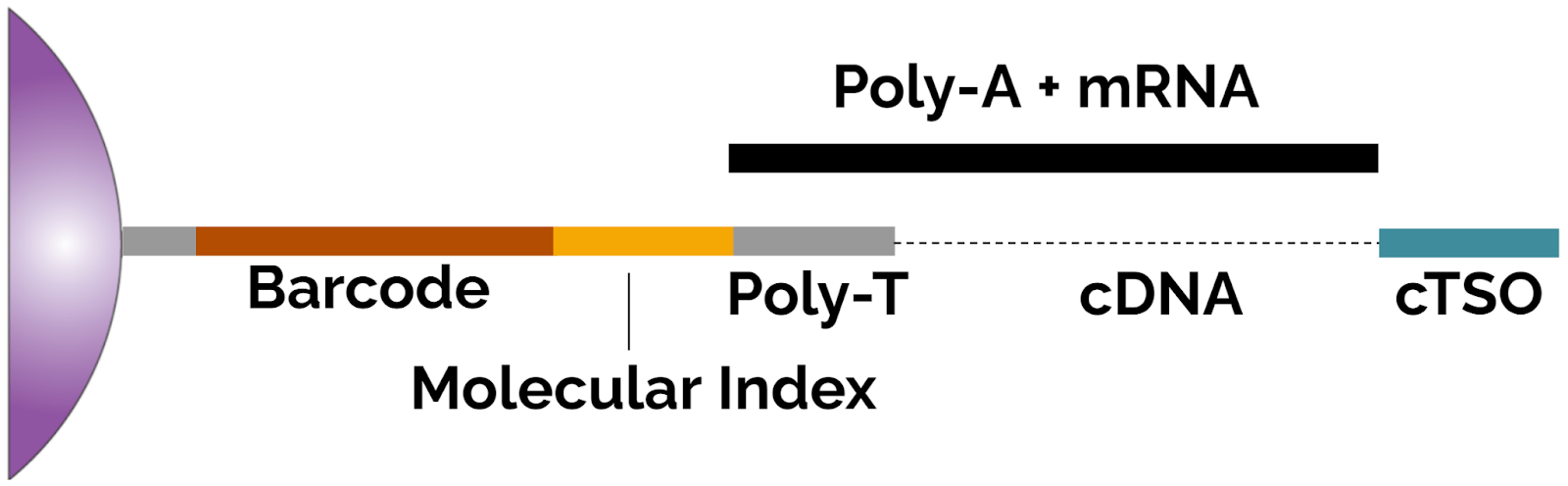
Data Analysis With Pipseeker Software Fluent Biosciences
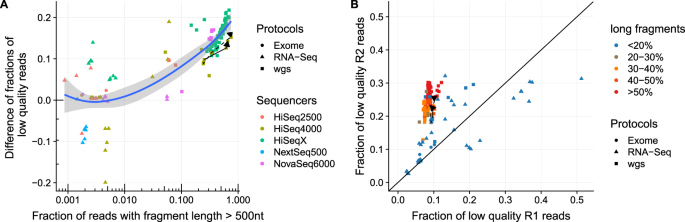
Long Fragments Achieve Lower Base Quality In Illumina Paired End Sequencing Scientific Reports

Paired End Sequencing The Inner Distance Between Paired Reads R1 And Download Scientific Diagram
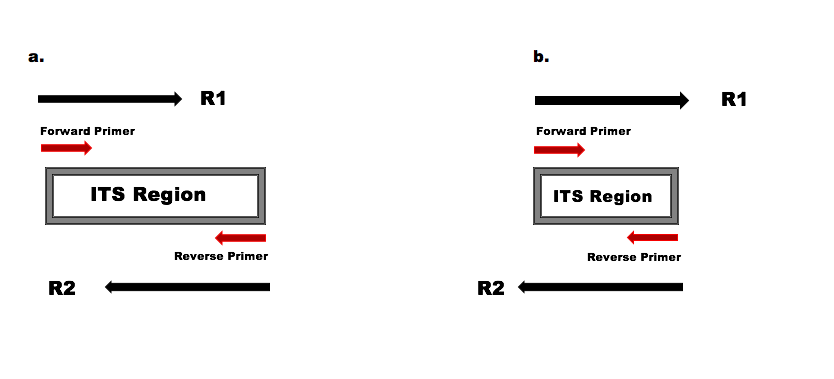
Dada2 Its Pipeline Workflow 1 8

Ngmerge Merging Paired End Reads Via Novel Empirically Derived Models Of Sequencing Errors Bmc Bioinformatics Full Text

Position Specific Averaged Mismatch Rate Of Short And Long Fragments In Download Scientific Diagram